 |
 |
|
| score: 3 | score: 2 |
Protein folding is a heavily studied question in biology: Given a chain of amino acids, how will the protein tend to arrange itself physically? The physical arrangement of the protein has important consequences for what the protein does in a biological system.
Computing the physical arrangement is very difficult. Some researchers instead examine a highly simplified model called the HP model, in which each amino acid appears on a grid, with connected amino acids placed at adjacent grid points. (This simplification is completely unrealistic, but the problem is still interesting.) We classify each amino acid into one of two categories — some are hydrophobic (represented with a capital H) and others are polar (capital P). The hydrophobic amino acids are non-polar and so despise being next to polar acids and water molecules. Thus, in a water-based solution, the protein will tend toward arranging the hydrophobic residues next to each other. In the HP model, then, we ask: How can we place the protein chain onto a grid so as to maximize the number of adjacent pairs of hydrophobic amino acids?
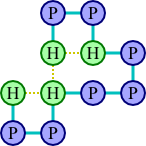
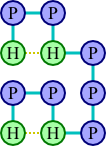
As an example, let's consider the protein chain HPPHPPPHPPH. Two possible ways of arranging this protein on a grid are illustrated below.
score: 3 score: 2
The arrangement at left has a score of three, since there are three pairs of adjacent hydrophobic amino acids (each diagrammed with a gold-dashed line), whereas the second has a score of two. The first arrangement would therefore be preferred. In fact, there is no arrangement with a score better than three, so the first arrangement is optimal.
Finding the optimal protein arrangement — even using the much-simplified HP model on two-dimensional grid — is NP-complete. But because the problem is important, researchers still work finding fast algorithms.
For this assignment, I have produced a program that uses exhaustive search to compute and display the optimal arrangement under the HP model. This code consists of two files (available in Java or Python).
Protein.javaprotein.pyDefines a class for representing a single protein-folding solution, including code to compute its score and to display it. Also, the class includes the mainmethod. You should not modify this file.Searcher.javasearcher.pyAll search code is contained in this file.
Your assignment is twofold.
First, you should optimize the program so that it computes the optimal arrangement more quickly. Your solution should still use exhaustive search, but you should strive to incorporate pruning and efficient computation to make it go more quickly.
This is an open-ended assignment: It will not be clear when your
work is good enough.
To give you an idea of the improvement you
should expect, I computed the optimal chain for the above HPPHPPPHPPH
example on my computer. The distributed dumb
solution took
400 ms; but after implementing three quite simple improvements,
the time dropped to 80 ms. That's not to say that you should be
satisfied by 80 ms — I expect you to be cleverer —
but if you're not see at least that much improvement, you should
probably keep working.
Second, you should write a brief paper describing the techniques that you employed to improve the program's performance. This paper should describe each of the improvements you made, and it should include some discussion of the effect of each improvement, including measurements. To measure the improvement, you should include results for about three protein chains.
The paper should be formatted as a simple ASCII text file, created using a text editor such as jEdit. The text of the paper should make sense without referencing your code implementation. Your assignment grade will depend in part on the quality of writing in this paper, including the quality of your algorithmic descriptions, the experimental technique, and your analysis.